
Définition
Description de la S.E.P
La sclérose en plaques est une maladie auto-immune qui affecte le système nerveux central. Elle entraine des lésions qui provoquent des perturbations motrices, sensitives et cognitives. A plus ou moins long terme, ces troubles peuvent progresser vers un handicap irréversible. Les traitements actuels permettent de réduire les poussées et améliorent la qualité de vie des patients, mais ils ont une efficacité insuffisante pour lutter contre la progression de la maladie. Cependant, de nouvelles stratégies thérapeutiques particulièrement prometteuses pourraient changer la donne dans les années à venir.
Une maladie auto-immune du jeune adulte
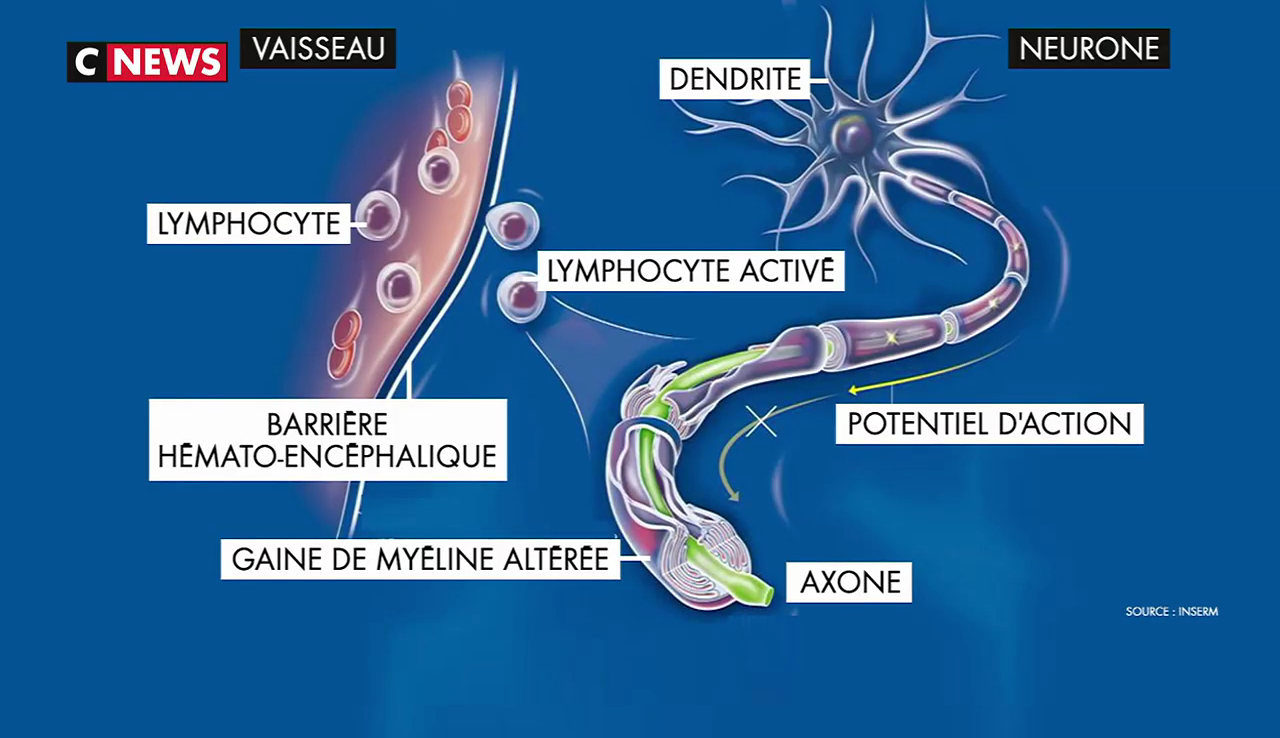
La sclérose en plaques (SEP) est une maladie auto-immune : le système de défense censé protéger le patient d’agressions extérieures, se retourne contre ses propres cellules et les attaque pour des raisons encore mal connues.
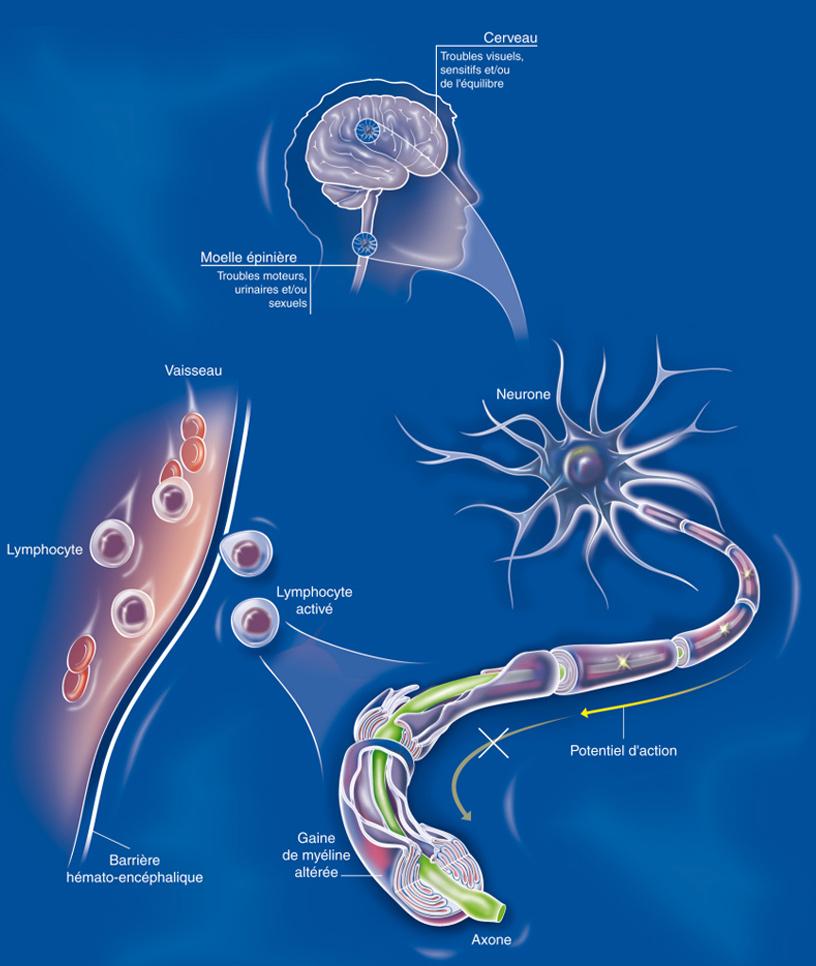
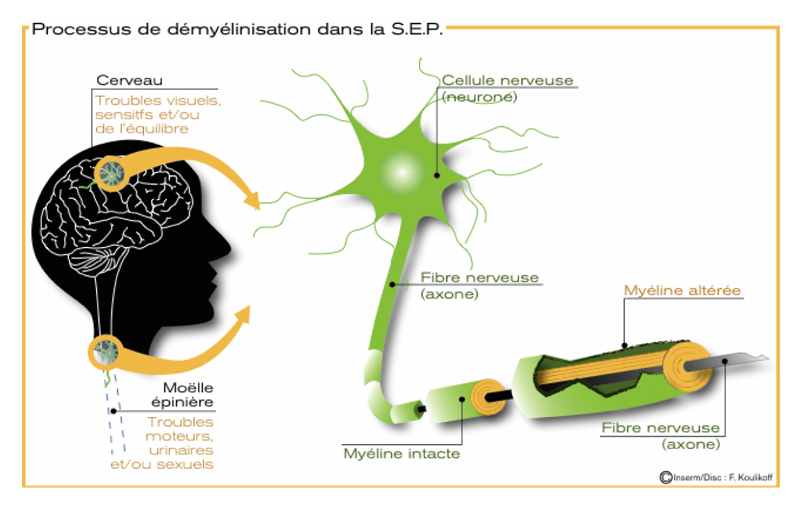
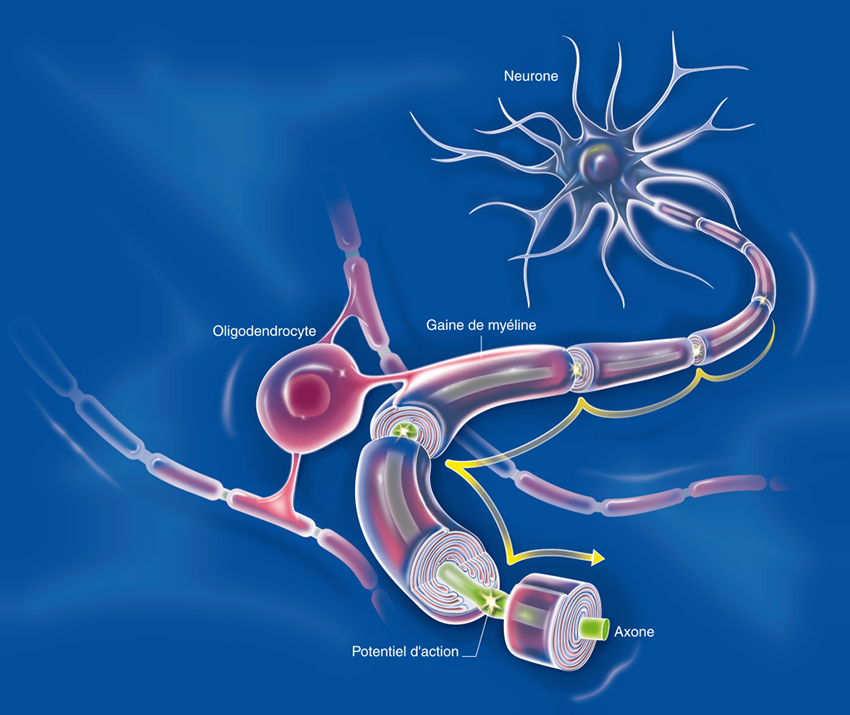
La maladie fait intervenir des mécanismes auto-immuns complexes qui attaquent les cellules chargées de synthétiser la gaine de myéline qui entoure les axones dans le système nerveux central. Ce phénomène entraine des lésions à l’aspect scléreux (épais et dur), dispersées dans le système nerveux central. Ces lésions sont appelées plaques, d’où le nom de la maladie. Elles traduisent une démyélinisation et souvent le début d’une dégénérescence axonale.
Qu’est ce que la myéline ?
La myéline est une membrane biologique qui s’enroule autour des axones pour constituer une gaine. Pour mémoire, un axone est le prolongement unique par lequel un neurone communique avec sa cellule cible.
La gaine de myéline sert à isoler et à protéger les fibres nerveuses comme le fait le plastique autour des fils électriques. Elle joue aussi un rôle dans la vitesse de propagation de l’influx nerveux transportant l’information le long des neurones. Deux types de cellules fabriquent la myéline : les cellules de Schwann dans le système nerveux périphérique et les oligodendrocytes dans le système nerveux central.
La sclérose en plaques est une maladie du jeune adulte qui représente la première cause de handicap sévère non traumatique chez les trentenaires. L’âge moyen de début des symptômes est en effet 30 ans. La maladie touche davantage de femmes, avec un sex-ratio de 1 homme pour 3 femmes environ. Environ 80 000 personnes sont touchées en France (environ 1 personne sur 1000).
Des facteurs de risque encore mal connus
La sclérose en plaques n’est pas une maladie héréditaire. Cependant, il existe des facteurs génétiques favorables à son développement, sous l’influence d’autres facteurs (notamment environnementaux). Ainsi, plusieurs membres d’une même famille peuvent être touchés.
La susceptibilité génétique
Une prédisposition génétique à la sclérose en plaques résulte de l’association de plusieurs variants génétiques, chacun d’entre eux ayant un faible effet sur le risque de développer la maladie. Une vaste étude d’analyse de génomes, impliquant deux consortiums de recherche internationaux (l’International Multiple Sclerosis Genetics Consortium et du Wellcome Trust Case Control Consortium), et notamment des chercheurs de l’lnserm, s’est achevée en 2011. Elle a permis de découvrir 29 variants génétiques associés à la maladie. La plupart de ces gènes jouent un rôle dans l’immunité. L’un d’entre eux est le gène HLA (Human Leucocyte Antigen, situé sur le chromosome 6) qui est impliqué dans la reconnaissance des cellules du « soi » par le système immunitaire. D’autres gènes codent pour des récepteurs de l’interleukine 2 et de l’interleukine 7, des médiateurs chimiques du système immunitaire.
Les facteurs environnementaux
Parallèlement à la prédisposition génétique, différents facteurs, notamment environnementaux influence le développement de la sclérose en plaques. Les facteurs climatiques, en particulier le niveau d’ensoleillement, sont les plus connus. En effet, la répartition de la maladie à travers le monde n’est pas uniforme : il existe un gradient de latitude Nord-Sud dans l’hémisphère Nord, et un gradient Sud–Nord dans l’hémisphère Sud, c’est à dire un gradient augmentant avec l’éloignement de l’équateur. On trouve ainsi des zones de haute prévalence de la maladie (supérieure à 100 pour 100 000 habitants) en Scandinavie, Ecosse, Europe du nord, Canada et Nord des Etats-Unis, des zones de prévalence moyenne (autour de 50 à 100) au Sud des Etats-Unis et en Europe centrale et de l’Ouest, et des zones de basse prévalence (inférieure à 5) autour de la Méditerranée et au Mexique. Toutefois, ce principe n’est pas généralisable à toute la population mondiale et la répartition de la SEP dans l’hémisphère sud n’est pas aussi schématique.
L’adolescence, période charnière ?
Des travaux portant sur des populations migrantes entre des zones de prévalence de la maladie différente suggèrent qu’il existe une étape clé au moment de l’adolescence. De façon très schématique, les individus qui migrent après l’âge de 15 ans conservent le risque de la région d’origine comme si celui-ci était inscrit dans les gènes. En revanche, ceux qui migrent avant l’âge de 15 ans acquièrent le risque de la région d’arrivée, comme si l’exposition environnementale au moment de l’adolescence déclenchait un événement décisif plusieurs années avant le début clinique de la maladie.
D’autres facteurs déclenchants sont soupçonnés comme le tabagisme actif ou le tabagisme passif au cours de l’enfance.
Il est important de noter que ces données reposent sur des observations, mais qu’aucun mécanisme expliquant comment ces facteurs participent au développement de la maladie n’a été identifié à ce jour. Pour l’heure, prévenir la survenue d’une sclérose en plaques chez une personne prédisposée est donc difficilement envisageable.
Les scientifiques cherchent depuis longtemps une origine infectieuse à la sclérose en plaques. Elle provoquerait un dérèglement du système immunitaire qui se mettrait à attaquer les gaines de myéline plutôt que les agents pathogènes. Cette hypothèse est très étudiée car d’autres affections proches de la SEP qui atteignent également la substance blanche, comme la leuco-encéphalopathie multifocale progressive et l’encéphalopathie subaiguë à VIH, sont d’origine virale. Dans le cas de la SEP, de nombreux virus ont été soupçonnés tour à tour : celui de la rage, de l’herpès, de la rubéole, de la rougeole, de la varicelle, certains rétrovirus comme le virus HTLV-1 responsable des leucémies de type T chez l’adulte, ou encore le virus Epstein-Barr, responsable de la mononucléose infectieuse. La bactérie Chlamydia pneumoniae a également été suspectée. Mais comme pour tous les virus cités précédemment, son rôle n’a jamais été prouvé. A ce jour, la piste infectieuse n’est donc qu’une hypothèse.
Pas d’imputabilité pour les vaccins contre l’hépatite B et le papillomavirus.
Des plaintes ont été déposées par des patients vaccinés contre l’hépatite B et, plus récemment en 2013, contre les papillomavirus impliqués dans des cancers gynécologiques. Plusieurs études ont été menées, y compris à l’Inserm, pour évaluer la sécurité de ces vaccins. Les résultats rassurants, faisant l’objet d’un consensus international, ont conduit les agences de santé à réaffirmer l’absence d’imputabilité de ces vaccins dans la survenue de la sclérose en plaques.
En ce qui concerne le vaccin anti-HPV, environ 5,5 millions de doses de vaccin avaient été distribuées fin 2013 et 503 cas d’effets indésirables graves, dont 17 cas de sclérose en plaques, avaient été rapportés sur la même période. L’Agence nationale de sécurité des médicaments et des produits de santé a mené une étude sur près de deux millions de jeunes filles et a constaté que le taux d’hospitalisation pour des maladies auto-immunes était le même chez les jeunes filles vaccinées que non vaccinées (environ 2 pour 10000 patients/année).
Une évolution imprévisible
La sclérose en plaques est une maladie extrêmement hétérogène d’un patient à l’autre, compte tenu de la variété des symptômes possible et de son évolution imprévisible. Le diagnostic est difficile car il n’existe pas de test diagnostic spécifique. Il repose sur un faisceau d’arguments, en particulier sur la dissémination de signes cliniques dans le temps et dans l’espace, associée à une inflammation limitée au système nerveux central.
Les symptômes varient beaucoup d’une personne à l’autre et se modifient aussi au cours de la vie chez une même personne. Dans 85% des cas, la maladie débute par des poussées (forme récurrente-rémittente). Les signes dépendent de la zone du cerveau ou de la moelle épinière touchée par les lésions. Il peut s’agir de troubles moteurs liés à une faiblesse musculaire, de troubles de la sensibilité, de symptômes visuels (vision double ou baisse d’acuité visuelle), de troubles de l’équilibre, de troubles urinaires ou sexuels… Ces différents signes cliniques peuvent être isolés ou associés. Ils peuvent survenir en quelques heures ou en quelques jours, et disparaître totalement ou partiellement en quelques semaines. Ces signes sont souvent associés à une fatigue extrême et inhabituelle, des troubles de la mémoire, de la concentration ou encore des épisodes dépressifs.
Les premières années, les symptômes disparaissent après chaque poussée et la récupération est souvent complète. Il peut s’écouler quelques mois ou plusieurs années entre deux poussées. Après un délai variable, de 5 à 20 ans, un handicap permanent peut s’installer et s’aggraver de façon progressive, avec ou sans poussées surajoutées : c’est la phase secondairement progressive de la maladie. Dans la moitié des cas environ, une difficulté majeure pour la marche est présente 20 ans après le début de la maladie.
Dans 15% des cas, la phase initiale de poussées (forme récurrente-rémittente) n’existe pas et les symptômes apparaissent directement progressivement puis s’aggravent avec le temps. Cette forme, appelée primitivement progressive, est de plus mauvais pronostic. Elle survient en général après l’âge de 40 ans
Des traitements qui modulent le système immunitaire
Les traitements disponibles à ce jour ne permettent pas de guérir, mais ils préviennent les poussées dans les formes récurrentes-rémittentes. Ils ne présentent pas d’efficacité sur les formes progressives de la maladie.
La cortisone est le traitement référence en cas de poussée inflammatoire et des traitements de fond sont utilisés pour moduler l’activité du système immunitaire.
Les immunomodulateurs sont les traitements de première ligne. Interféron β et acétate de glatiramère atténuent l’activation des lymphocytes T, inhibent la production de cytokines Th1 pro-inflammatoires, activent la sécrétion de cytokines Th2 anti-inflammatoires. Ils diminuent d’environ 30% la fréquence des poussées et réduisent d’environ 60% le nombre de nouvelles lésions visibles à l’IRM. De nouveaux immunomodulateurs par voie orale seront commercialisés très prochainement : le diméthyl fumarate et le teriflunomide.
Les immunosuppresseurs sont des traitements plus agressifs, utilisés en seconde ligne pour prévenir l’apparition des poussées. Leur utilisation est limitée aux formes sévères et très actives de la maladie en raison de leurs effets indésirables potentiellement graves. Ils entrainent une déplétion en lymphocytes B et/ou T via différents modes d’action. Parmi eux, l’anticorps monoclonal natalizumab diminue la fréquence des poussées d’environ 60% et le nombre de nouvelles lésions à l’IRM d’environ 90%. Il diminue d’environ 40% le risque d’aggravation du handicap neurologique à deux ans. Le fingolimod, disponible depuis plus d’un an, réduit d’environ 50% la fréquence des poussées. Il est aussi évalué dans les formes progressives de la maladie.
Parmi les molécules en phase de développement avancée, l’alemtuzumab a permis de réduire de moitié le risque de poussée, par rapport à l’interféron bêta (essais de phase 3). Le daclizumab (anti-récepteur de l’interleukine 2) et l’ocrelizumab (qui cible les lymphocytes B) ont aussi donné des résultats intéressants. Ce dernier traitement est en développement dans les formes rémittentes et primitivement progressives de la maladie. Obtenir une meilleure sécurité d’emploi, ainsi qu’une efficacité dans les formes progressives de la maladie sont deux objectifs majeurs attendus avec les thérapeutiques en cours de développement.
Ces nouveaux médicaments augmentent la complexité de la prise en charge des patients : les traitements diffèrent par leur voie d’administration, leur mécanisme d’action, leur efficacité, mais aussi leur sécurité d’emploi. En outre, tous les patients ne répondent pas de la même façon aux traitements. Des études sont en cours afin de comprendre ces différences et identifier des marqueurs prédictifs de la réponse aux traitements, de manière à pouvoir optimiser la prise en charge et le choix des médicaments. Dans cet objectif, l’Observatoire français de la sclérose en plaques a mis en place un programme (EDMUS), comprenant en particulier une base de données relatives au suivi de 40 000 patients.
En parallèle aux traitements médicamenteux, la rééducation est un aspect important de la prise en charge des patients atteints de sclérose en plaques. Elle est indispensable à chaque stade de la maladie, pour l’entretien musculaire, pour éviter des complications liées à l’immobilité ou pour permettre au patient de poursuivre des activités quotidiennes.
Vers de nouvelles stratégies immunomodulatrices et la remyélinisation
De nouvelles stratégies immunomodulatrices sont aujourd’hui envisagées. Des chercheurs tentent par exemple de rendre le système immunitaire tolérant aux cellules qui produisent la myéline (les oligodendrocytes) en l’exposant progressivement à des antigènes myéliniques exogènes. Des travaux conduits sur des modèles animaux ont donné des résultats encourageants avec l’administration d’antigènes très spécifiques.
Une autre approche est centrée sur l’hypothèse rétrovirale. L’implication dans la sclérose en plaques d’un rétrovirus endogène (multiple sclerosis-associated retrovirus, MSRV) est discutée depuis de nombreuses années. Des travaux suggèrent que la protéine d’enveloppe du virus pourrait activer une cascade proinflammatoire. Ces travaux ont conduit à la mise en œuvre d’un essai clinique utilisant un anticorps monoclonal dirigé contre cette protéine d’enveloppe.
Des stratégies complémentaires à l’immunomodulation et à l’immunosuppression connaissent en outre actuellement un engouement : il s’agit de la remyélinisation et de la neuroprotection. Elles pourraient permettre de freiner l’évolution de la maladie et l’apparition du handicap associé.
La remyélinisation, un phénomène parfois spontané
L’existence d’une remyélinisation spontanée est connue depuis plusieurs décades. Elle est souvent restreinte à la périphérie des plaques mais peut parfois être complète (ce sont les « shadow plaques », appelées ainsi car elles fixent moins le colorant myélinique, d’où leur aspect « ombré »). Environ deux tiers des lésions seraient ainsi réparées, partiellement ou en totalité. Toutefois, tous les patients ne sont pas égaux devant ce phénomène. Les progrès de l’imagerie médicale, notamment l’utilisation de traceurs se fixant sélectivement sur la myéline et observables en tomographie par émission de positons, permettent en effet désormais d’évaluer le degré de remyélinisation chez des patients. Il s’agit pour l’instant d’une procédure expérimentale, développée dans le cadre d’une collaboration entre l’Inserm et le CEA.
La remyélinisation peut être stimulée par voie endogène ou exogène. Par voie endogène, il s’agit de provoquer la réparation spontanée de la myéline. Les mécanismes cellulaires et moléculaires impliqués dans cette remyélinisation à partir des précurseurs des oligodendrocytes sont de mieux en mieux connus. Cela a conduit à l’identification de facteurs activateurs et inhibiteurs. Lingo-1 est l’un de ces facteurs inhibiteurs : son expression par les cellules oligodendrogliales immatures empêche leur différenciation. Dans des modèles expérimentaux, le blocage de Lingo-1 favorise la myélinisation et la remyélinisation. Un anticorps monoclonal anti-Lingo-1 humanisé est en cours de développement (deux études de phase 2 en cours).
La seconde voie est la remyélinisation par voie exogène, c’est-à-dire la greffe de cellules myélinisantes. L’objectif est de recréer des oligodendrocytes producteurs de myéline dans les lésions, en injectant des cellules souches. Chez l’animal, l’injection de cellules souches neurales favorise la remyélinisation à partir de cellules endogènes. Il se pourrait donc que les cellules greffées agissent en sécrétant des facteurs neurotrophiques nécessaires à la production endogène de myéline.
Pour cette dernière approche, il est nécessaire de guider les cellules greffées vers des sites de lésion. Pour cela les chercheurs tentent d’identifier des molécules de guidage. C’est le cas des molécules de la famille des sémaphorines, impliquées dans le guidage de la migration des précurseurs d’oligodendrocytes au cours du développement. Ces sémaphorines sont ré-exprimées dans le système nerveux de patients atteints de sclérose en plaques et pourraient servir à attirer les cellules précurseurs des oligodendrocytes vers les zones lésées. A l’inverse d’autres molécules empêchent ce recrutement, comme la nétrine, inhibant ainsi la remyélinisation.
Enfin, d’autres chercheurs s’intéressent au microbiote (flore intestinale) des patients dont la composition jouerait un rôle dans l’inflammation et la régulation du système immunitaire. Cela semble établi pour certaines maladies auto-immunes et pourrait aussi se vérifier pour la sclérose en plaques.
[ source INSERM ]
Descriptif complet (& précis) de la sclérose en plaques

Ce document .powerpoint décrit en détail la maladie :
- Epidémiologie
- Anatomo-pathologie
- Etiopathogénie
- Clinique
- Atteinte pyramidale
- L’atteinte sensitive
- Atteinte cérébelleuse
- Névrite rétrobulbaire
- Atteinte vestibulaire
- Atteinte du tronc cérébral
- Atteinte oculomotrice
- Troubles psychiques
- Troubles sphinctériens et génitaux
- Manifestations paroxystiques, etc..